Liver Illness
At a glance
- Wilson’s disease is caused by an accumulation of copper in the liver, kidneys, heart and eyes
- Symptoms may include jaundice, abdominal discomfort and personality changes
- While it affects men and women equally, females are more likely to experience acute liver failure
What is it?
Copper is a mineral that is important for our general health, but too much of it is toxic. Because we only need tiny amounts, our bodies get rid of excess copper in bile. Wilson’s disease is a genetic or inherited condition, in which our bodies cannot get rid of this excess copper, and it consequently builds up in the liver, brain and other organs. The copper typically builds up in the cells of the liver first, as this is where this mineral is generally stored before excretion. Here, it causes destruction of liver cells which causes inflammation and liver fibrosis (scarring) and in some instances, cirrhosis (permanent scarring which interferes with liver function). When the liver is damaged, excess copper then accumulates in the brain, kidneys, joints, heart and eyes.
Typical symptoms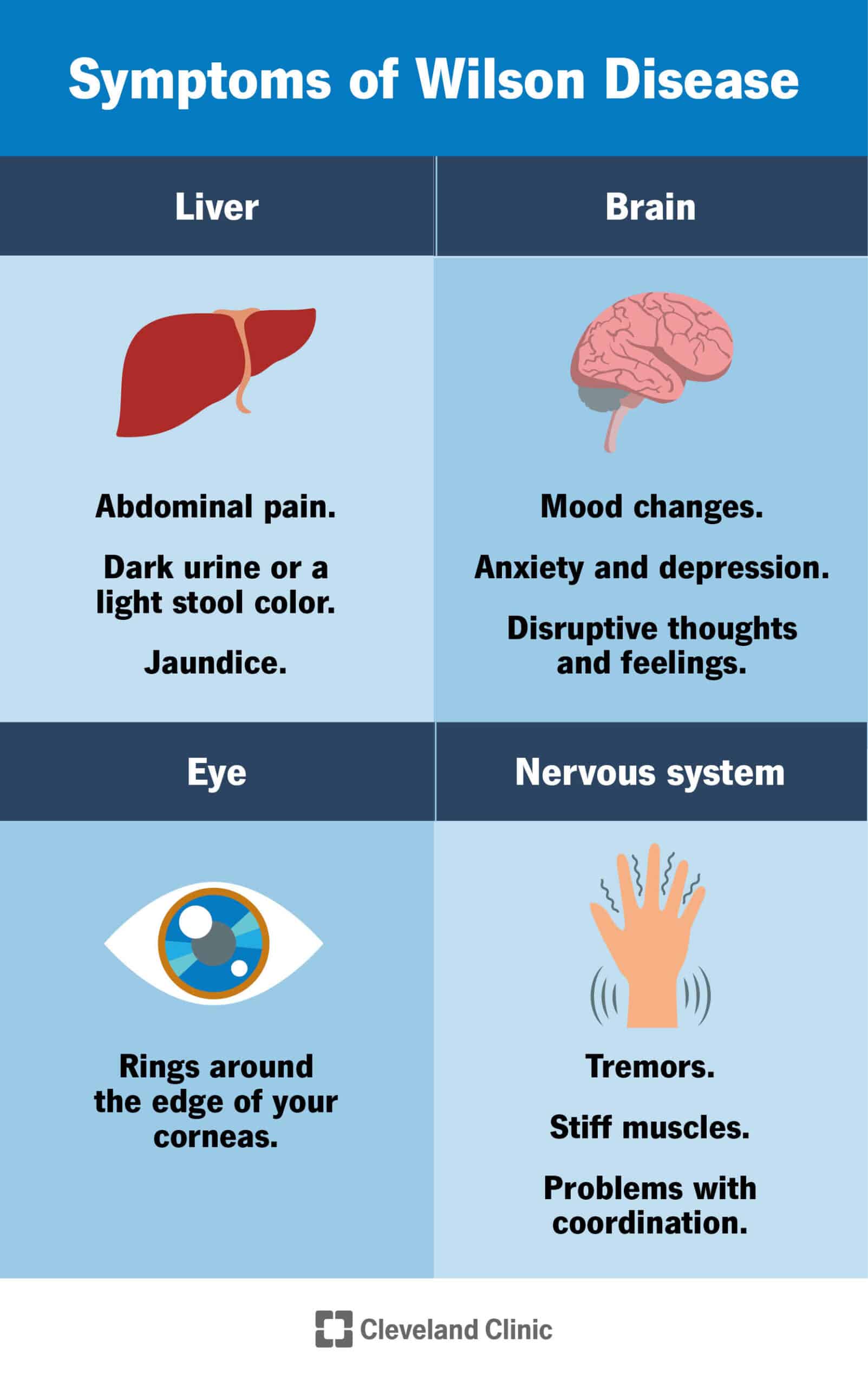
Since copper typically accumulates in the body from birth, symptoms may be first displayed at any time from six to 40 years, but most commonly in the late teens. Symptoms such as jaundice, abdominal discomfort or pain, ascites (bloating and swelling of the abdomen with fluid) or vomiting of blood might occur. Clinical presentation of an individual with Wilson’s disease might include elevated liver function tests (LFTs) or blood liver enzymes, hepatitis (liver inflammation) or liver fibrosis or cirrhosis. Others might be tremors, difficulty speaking, personality changes, clumsiness or loss of muscle control.
Diagnosis
As Wilson’s disease is an inherited genetic condition (caused by a mutated or faulty gene called ATP7B), genetic testing might be the most robust diagnostic technique, since the liver and neurological symptoms displayed may not be specific enough to differentiate Wilson’s disease from other liver diseases, or from a psychological illness. Additionally, a rusty or copper coloured ring around the eye of an individual with Wilson’s disease might also contribute to a diagnosis. Known as Kayser Fleischer rings, an ophthalmologist will diagnose this phenomenon.
Incidence
It takes two faulty or mutated genes for an individual to develop Wilson’s disease – one from the father and one from the mother. While many people may be carriers of the affected gene (one in 100 people) it is a very rare disease with an incidence of one in 30,000 of the population. This disease affects men and women equally, but females are more likely to experience acute liver failure.
Treatment
While there is no cure for Wilson’s disease, treatment, regular monitoring and diet can play a role in its management. Treatment aims to remove excess copper from the body and prevent it from building up to harmful levels again. Chelating agents, which bind to copper and ensure it is excreted in urine, are typical lifelong medications given to individuals diagnosed with Wilson’s disease. They might also be encouraged to maintain a low copper diet, excluding food such as shellfish, wholegrains, beans, nuts, dried fruit, yeast and potatoes.
Prognosis
Diagnosed and treated in the early stages of the disease, and prescribed lifelong chelating agents, one can anticipate an active, healthy and normal life, living with Wilson’s disease.